
混合系统的数据完整性
随着《药品记录与数据管理要求(试行)》 的实行,国内医药研发和生产单位也更加重视数据完整性的要求,开始重新购置计算机化系统和相关仪器设备——一些老旧系统和设备无法满足数据完整性对计算机化系统的要求,企业不得不采用增加纸质记录的方式弥补计算机化的缺陷,从而产生了混合系统。在实践中,相对纸质系统和计算机化系统来说,混合系统的高风险愈发清晰,那么如何识别和控制这些风险就变得迫切起来。工作中,大家会产生各种关于混合系统的疑惑,例如,什么样的系统才是混合系统?能否用纸质文件进行签字审批?纸质记录可不可以代替电子记录?同时,基于混合系统的高风险性,各国药监部门高度关注混合系统,相关的审计缺陷也较为常见,如Able 实验室的审计缺陷[2]。本文将结合各国药监部门的数据完整性法规和行业最佳实践探讨混合系统数据完整性的风险管理与控制。
1 混合系统的定义
相关药监部门和行业组织从不同的维度给出了混合系统的定义。
1.1 WHO关于混合系统的定义[3]
混合系统:使用电子系统和纸质系统的联合体。
1.2 PICS的定义[4]
混合系统:一种管理和控制数据的系统,通常由一个电子系统组成,但需要一个预定义的手工系统加以补充。混合系统依赖于两个子系统的有效管理来实现正确的运行。
1.3 MHRA的定义[5]
混合系统:纸质记录和电子记录共同构成的原始记录。
1.4 ISPE的描述[6]
混合情况:纸质记录/ 签名和电子记录/ 签名共存的情况。
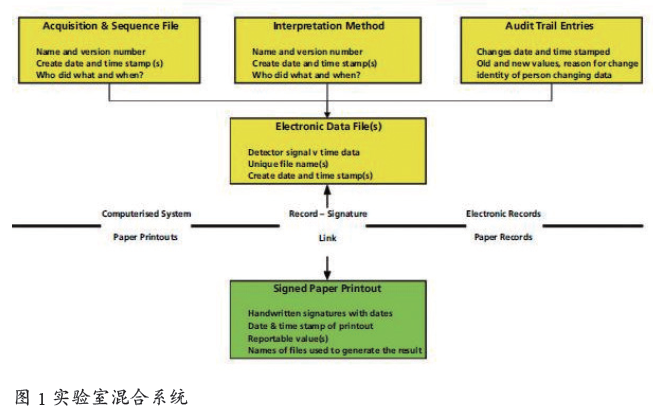
综上所述,混合系统可以理解为:计算机化系统(电子记录)与纸质系统( 纸质记录) 的组合——以共同实现GxP 业务。实验室混合系统[7] 如图1 所示。
2 混合系统的相关法规要求
2.1 国家药品监督管理局在《药品记录与数据管理要求(试行)》中的要求[1]
第四条:“ 记录载体可采用纸质、电子或混合等一种或多种形式。”
第六条:“对于电子记录和纸质记录并存的情况,应当在相应的操作规程和管理制度中明确规定作为基准的形式。”
同时在第四章和第五章中详细描述了对计算机化系统的要求,例如,对时间控制、权限访问控制和审计追踪的要求。国家药品监督管理局在法规中就考虑到了实践中混合系统的应用情况,但没有针对混合系统提出具体要求,而是分散在相应的纸质记录和计算机化系统的要求中。
2.2 欧盟GMP的要求[8]
许多文档(指南/ 记录)或许存在混合格式,有些元素是电子格式,有些元素是纸质记录。需要针对混合系统建立关联与控制措施。应有相应的控制措施以保证记录在整个生命周期内的完整性。
2.3 WHO数据完整性指南的要求[3]
如果计算机化系统仅支持单个用户登录或有限的用户登录,并且没有合适的系统可替代计算机化系统,则应通过第三方软件或提供可追溯性(含版本控制)的纸质方法提供等效的控制措施。应证明并记录替代系统的适用性。不建议使用传统的混合系统,并应确定升级/ 替换的优先级时间表。
2.4 PIC/S以及GMP/GDP环境数据管理和完整性的良好实践[4]
PIC/S在9.8节混合系统的管理中特别强调:
混合系统需要具体的和额外的控制,以反映其复杂性和对数据操纵的潜在脆弱性。
混合系统的每个组成元素都应按照上述有关手工和计算机系统的指南进行确认和控制。
混合系统通常由计算机系统和手工系统组成。应特别注意确认:
● 计算机化系统的确认/ 验证范围;
● 由于难以一致地应用手工过程,因此应用于混合系统的手工元素管理应具有控制系统鲁棒性。
2.5 FDA CFR 21 Part 11电子记录与电子签名[9]
对电子记录采用的电子签名或手写签名,必须保持其签名与记录的关联关系,确保签名不能被轻易地删除、复制或转移,避免数据做假。手写签名关联到电子记录即属于典型的混合系统。
2.6 FDA数据完整性与cGMP遵从的行业指南问答[10]
在问题10/11/12 中,FDA 回答了静态数据与动态数据的保存以及相关的签名问题。在很多情况下,混合系统无法满足法规的要求,特别是动态数据。
2.7 FDA cGMP2级指南记录与报告[11]
问题3回答并解释了当前行业对指南(电子签名与电子记录范围与应用)的一个误解,即在任何情况下都可以使用打印的电子记录作为电子记录的真实副本来进行相关的GxP活动。FDA以HPLC和GC为例解释说明了色谱图的打印版并不是真实副本,不能满足法规的要求。
2.8 ISPE GAMP指南记录与数据完整性[6]
在章节19.2 混合系统中,描述了混合系统的控制措施并举例说明了混合系统在实践中遇到的困难。
2.9 PDA TR80制药实验室的数据完整性管理体系[12]
在章节6.2 混合系统中,基于法规要求和混合系统的原生风险,PDA 识别了相关的风险点并给出了具体的控制措施。
综上法规与指南的描述,由电子记录和纸质记录组成的混合系统,要求更详尽的风险分析和更为严格的控制措施。
3 混合系统的特点与表现形式
一般说来,由于计算机化系统的缺陷(如缺失访问控制、审计追踪等),企业不得不额外采用纸质记录的方法来弥补计算机化系统的缺陷(如采用日志的方式代替审计追踪),混合系统由此而生。
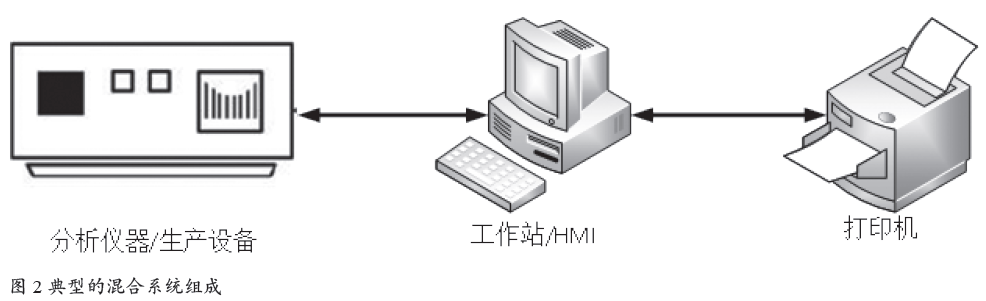
3.1 典型的混合系统组成
典型的混合系统组成如图2所示。
3.2 常见混合系统的类型
3.2.1 计算机化系统有用户控制但无审计追踪功能
常见的例子有:早期购买的实验室红外、紫外仪等,早期购买的反应釜、混合机等生产设备,以及主要为其他行业设计的实验室仪器和生产设备,如简单PCR仪、蛋白质凝胶系统等。
3.2.2 计算机化系统无用户控制也无审计追踪功能
计算机化系统有用户控制,但必须使用共享操作系统用户名,如共享Windows 管理员用户名登录操作系统。常见的例子:有些简单仪器的应用程序,其软件设计要求调用Windows 管理员用户。Administrator 的权限导致分析化验工作人员必须使用Administrator 登录操作系统才可以正常工作;有些包装机的HMI系统在开机时,系统使用默认的WinCC 管理员用户Administrator登录操作系统,并自动跳转到应用程序的操作界面。
计算机化系统共享用户名登录应用程序进行工作。常见的例子:有些生产设备只提供了有限的用户登录权限;有些设备工艺时间超过一个班次,登录用户启动工艺后便不可退出应用程序(中途退出应用程序将会导致数据丢失),导致后续班次继续使用前面班次的登录用户名,即使应用程序有完善的用户权限管理功能也无法改变这一状况。
3.2.3 计算机化系统有用户控制和审计追踪功能,但是存储容量有限
常见的例子:使用有限存储容量的冻干机和有限存储容量的小型分析仪器。
3.3 补救措施
基于上述功能缺陷,常见的补救措施是建立纸质日志流程,通过操作日志的方式尽可能地弥补用户权限/ 审计追踪的缺失。
4 混合系统的控制
4.1 混合系统带来的风险
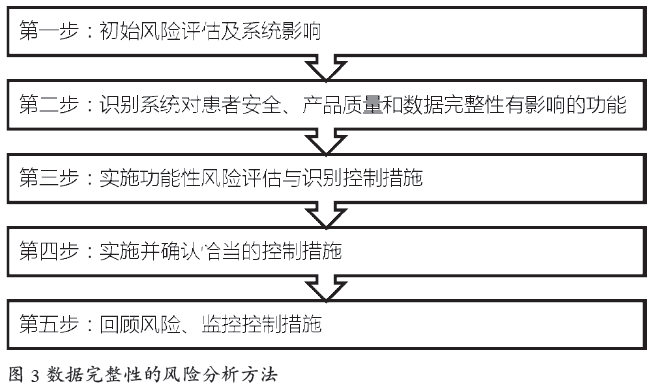
ISPE给出了数据完整性的风险分析通用方法[6],如图3所示。
风险分析需要结合具体的系统和业务实践,实践中常见的混合系统风险如下:
● 共享用户名,用户授权冲突;
● 数据缺乏可追溯性(缺乏密码控制,无审计跟踪/ 故意禁用审计跟踪,以及日期和时间易受手动更改影响);
● 审阅者难以实质性地审阅数据,包括元数据和审计追踪;
● 测试直到合格或重复打印;
● 低劣的备份恢复功能或单点故障(硬盘驱动器);
● 难以保留源电子数据和“完整”且包含所有元数据的数据;
● 电子数据与纸质签名难以同步。
4.2 混合系统常见控制措施
由于混合系统的风险来自于计算机化系统自身功能的不完备,导致很难找到技术方面的控制措施,更多的是从流程和人员素养方面寻找方案。
常见的控制措施有:
● 用户授权的管理:限制修改操作系统时间的权限;限制使用管理员用户登录操作系统;限制访问特定文件夹的权限,如存放关键数据的文件夹,或存放工艺数据的文件夹;限制删除回收站文件的权限,以免蓄意删除文件;限制U盘的使用权限等;
● 变更控制:计算机化系统和纸质记录的变更也需要遵从企业内部的变更流程;
● 审计追踪:因为不存在严格意义上的由应用程序自动生成的审计追踪记录,而是替代性地采用纸质日志记录的方式来追踪,因而需要更好地维护纸质审计追踪与相应的电子记录之间的关系,也需要更频繁和更严格的纸质审计追踪审阅;
● 流程控制:对于混合系统,企业应建立电子数据及相应的纸质记录的审阅与签字流程,包括纸质审计追踪的审阅;电子数据及相应的纸质记录的保存期限及保存期内的可读性检查:数据的保存应保持用户、时间、审计追踪等元数据的完整;混合系统的使用培训。
尽管我们可以从技术、人员、流程方面给出一些弥补措施来降低混合系统的风险,但实践中仍然存在巨大的困难。例如,通过纸质日志的方式来实现审计追踪,但仍然存在问题:
● 如何保证所有的操作都被如实的记录在日志中?
● 当日志和电子记录的数量急剧增加后,如何保持电子记录与相应的纸质审计追踪之间的关联关系?以及如何能快速的找到这种关联关系?
尽管在理论上,制药企业可以解决上述问题,但实践上为解决上述问题所带来的巨额人力物力支出令企业难以负担。
5 结语
本文讨论了混合系统的特点与现状,也总结了监管部门的要求,并给出了相应的风险控制措施。但从监管部门和实践的角度来看,混合系统并不能合规,停用混合系统并升级到满足合规要求的计算机化系统才是更好的解决措施。
【参考文献】
[1] 国家药品监督管理局,《药品记录与数据管理要求(试行)》,2020.
[2] Able Laboratories Form 483 Observations,6 July 2005; Available from: http://www.fda.gov/downloads/aboutfda/centersoffices/offic eofglobalregulatoryoperationsandpolicy/ora/oraelectronicreadingroom/ucm061818.pdf.
[3] WHO TRS 1033, Guideline on data integrity,2021.
[4] PIC/S PI041-1(draft 3)GOOD PRACTICES FOR DATA MANAGEMENT AND INTEGRITY IN REGULATED GMP/GDP ENVIRONMENTS,2018.
[5] MHRA,‘GXP’ Data Integrity Guidance and Definitions,2018.
[6] ISPE GAMP Guide: Records and Data Integrity,2017.
[7]R. D. McDowall, Data Integrity and Data Governance Practical Implementation in Regulated Laboratories,2019
[8] EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, 2011.
[9] FDA,21 CFR 11 Electronic Records; Electronic Signatures, Final Rule, in Title 21, 1997.
[10] FDA Draft Guidance for Industry Data Integrity and Compliance with cGMP, 2016.
[11] FDA Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance - Records and Reports.,2016.
[12] PDA Technical Report No.80 Data Integrity Management System for Pharmaceutical Laboratories,2018.



![]()
2001-2009Vogel Industry Media版权所有 京ICP备12020067号-15 京公网安备110102001177号

