
无菌检查试验的先天缺陷及应对策略的思考
为了最大程度降低药典无菌检查试验方法缺陷造成的影响,规范我国药品标准中无菌检查试验方法。借鉴西方国家对无菌检查法的管理经验,结合我国实际情况,探讨及规范我国药品标准中无菌检查法的具体措施。表明我国药典或相关法规应加强对无菌检查试验的原理、不足和注意事项等的解释说明,对生物制品的无菌检查方法应有专门规定。
无菌检查试验最早于1932年出现在英国药典中,从诞生到现在的几十年里,它作为产品质量检测试验一直存在广泛的争议。直到最近,美国药典、欧洲药典和日本药典的代表组成的药典讨论组完成对三方药典中无菌检查试验的协调统一,无菌检查试验的几个先天性缺陷也没有被完全克服。
为了消除这些缺陷对试验结果带来的不利影响,一些国家的管理部门和有关国际组织相继发布了对协调后无菌试验的说明和改进的文件。我国现行药典中无菌检查试验方法与欧美药典大体相似,在将来发布的2015年版药典与欧美药典将更趋于一致,无菌检查试验的这些缺陷无疑同样也会对我国药品生产和管理带来影响,分析和借鉴西方国家的经验教训是规范我国无菌检查试验的重要方式。
本文目的是通过分析西方国家和相关国际组织规范无菌试验的做法,探讨我国规范和优化无菌试验需采取的相应措施。
1.无菌检查试验的先天缺陷
采样
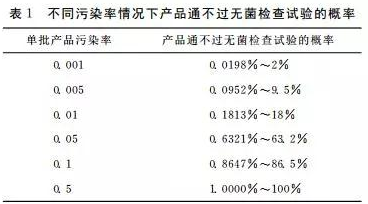
由于没有从统计学的角度考虑采样,药典中的无菌检查试验效率低。样品量少,使无菌检查试验检测污染物的能力受到限制,污染程度越轻,出现错误结果的可能性越大。有学者从统计学角度阐述了这个问题:①λ为单件产品受污染的可能性;②按泊松分布,从总体中抽到一个无菌样本的概 率 为e-λ(e为自然对数的底,约等于2.718281);③如果从一个假设为无穷个包装单位的药品单批中抽取20个样品;④ 通过无菌检查试验的概率为P20,通不过无菌检查试验的概率为1-P20,得出在不同污染率的情况下产品通不过无菌检查的概率(见表1),从表中可见,污染率越低对污染物漏检的可能性越大。由于不可能对一批成品的每一包装单位进行检测,所以,没有绝对的无菌保证。药品生产者必须遵守现行药品生产良好质量管理规范(CGMP)、无菌生产工艺的验证、良好厂房设计、员工的培训,才能取得高水平的无菌保障。
培养条件
研究表明,药典规定的微生物培养条件并不是许多细菌和真菌的最适生长条件。药典中胰酪胨大豆胨肉汤培养基在20~25 ℃条件下培养,目的是检测环境中的污染菌,许多临床上有意义的细菌生长在25~40 ℃之间,真菌生长在25~30 ℃之间。有培养基生产商推荐胰酪胨大豆胨肉汤培养基用于药典要求之外时的培养温度是35±2℃。美国药典无菌试验专家委员会推荐25~30℃为细菌、酵母菌和霉菌的培养温度。用7个不同的培养基检查88株不同的细菌、38株酵母菌和54株霉菌,结果发现,在32 ℃ 条件下对需氧菌和厌氧菌进行培养,胰酪胨大豆胨肉
汤培养基和硫乙醇酸盐流体培养基的效率低于亚硫酸钠-巯基乙酸盐肉汤;26℃培养真菌和酵母菌10天后,胰酪胨大豆胨肉汤培养基的效率不及沙氏液体培养基、沙氏葡萄糖琼脂、蛋白胨肝消化肉汤和蛋白胨肝消化琼脂。胰酪胨大豆胨肉汤培养基不适用于对苛求微生物(如嗜血杆菌或奈瑟氏菌属)的培养。硫乙醇酸盐流体培养基也不适用于苛求厌氧菌 (如普雷沃氏菌属)的恢复培养,培养基中需加入高铁血红素和维生素K1。
各国药典规定的培养时间为14天。美国药典和欧洲药典分别于2000年和1998年将原先培养7天的试验修改为14 天, 目的是关注那些由于“ 生长缓慢”及7天内可能无法检测到的微生物。微生物检测试验14天以上的试验,对于制造商来说是一个负担,不利于企业及早采取纠正措施。
2.西方国家相应的对策
为克服药典无菌检查试验先天缺陷带来的不利影响,西方国家在药典或相关法规中采取了相应的对策。
美国
2012年修订的美国联邦法规21CFR610.12,规定了一个不同于药典描述的无菌检查方法(CBER方法),适用于美国食品药品监督管理局生物制品审评和研究中心职权管理范围内生物制品的检测。FDA修订该法规的目的是为生物制品制造商提供更大的灵活性,以确保生物制品的安全性,并鼓励使用最合适和最先进的测试方法。
21CFR610.12取消了原有的细节上的规 定,如微生物、培养基、培养时间、培养温度、取样量和采样计划等内容,使用命令语气 “ 必须”从原则上强调无菌检测的要求。关于采样,至少要考虑:①被检测成品批的数量和体积;②生产持续时间;③最终包装的形态和大小;④ 可能的抑制剂、中和剂、防腐剂的量或浓度;⑤导致样品稀释的检测材料的量。该法规也允许使用非培养的检测方法。
欧洲
在 “2.6.1无菌检查” 之外,欧洲药典的非强制章节 “5.1.9无菌试验使用指南”向读者详细解释了 无菌试验的目的、存在的缺陷及注意事项等。该章指出,无菌试验合格结果的保证水平取决于批次的均一性、生产条件、采样方法等的有效性;对于最终灭菌产品提供灭菌过程的证据比无菌试验更重要。
2007年新载入欧洲药典的 “细胞产品的微生物控制”指出,本节描述的方法对于某些细胞产品来说比 “ 无菌检查”更快、更敏感。在专论中指定使用该方法替代 “ 无菌检查”的细胞产品。该节没有对所使用的培养基作具体要求,只是从原则上要求培养基要适合真菌、需氧菌和厌氧菌的生长,作为举例提出血培养基。培养温度35~37 ℃,高于三方协调后无菌检查方法。培养时间不超过7天或14天。
澳大利亚
澳大利亚卫生及老年保健部治疗产品管理局,在2006 年发布的《治疗产品无菌检查指南》中,对协调后的药典无菌检查方法做出了全面详细的解释说明,同时,还提出了控制无菌检查试验的附加试验-停滞试验 (StasiaTest )。澳大利亚执行英国药典,其无菌检查方法与欧洲药典一致。《 治疗产品无菌检查指南》的原理部分指出:① 统计学考虑也是无菌检验成功的重要一环;②药典无菌检查方法不利于对低水平污染的检出;③绝对的结果需要用能支持所有可能污染菌生长的培养基对每种产品进行检验。与协调后的药典无菌检查方法相比,该文件对培养基有如下两个新规定。
1.停滞试验
停滞试验不是药典的强制试验项目。其目的是要证明使用的培养基在整个培养期是否具有支持生长的能力。例如证明硫乙醇酸盐流体培养基能保持厌氧状态以满足生长缓慢的厌氧菌生长。
2.使用培养基
一般使用胰酪胨大豆胨肉汤培养基和硫乙醇酸盐流体培养基,但是,当产品特性或制造方式可能导致苛求微生物存在时 (如疫苗、血液制品等),使用替代的培养基是可行的,必须验证替代培养基有能力支持产品中可能出现的微生物的生长。
3.对规范我国药品标准中无菌检查试验的思考
关于培养时间过长的问题,中国药典与美欧药典都有鼓励使用快速微生物替代方法的验证指导原则。除此之外, 我国药品标准中还应体现如下内容。
应对无菌检查试验做出充分的解释
我国少数药品生产企业对药品GMP存在认识和重视程度不足的问题,部分从业人员对药品无菌检查试验的原理和作用也存在理解不深的问题,导致无菌检查试验 法不规范,上市药品还存在微生物污染事件的发生。药品监督管理部门对生产企业加强监管的同时,对药典等药品标准中无菌检查试验的进一步规范和解释也是促进我国药品质量提高的重要步骤, 如制订与 欧洲药典“无菌试验使用指南”相类似的文件。
我国药典对无菌检查试验的具体方法做了详细而严格的规定,但是,没有对无菌检查试验的 原理、先天缺陷、所起作用等做详细的解释说明,只提出了应该怎样做,没有说明为什么这样做。这样导致的一个后果是无菌检查结果的滥用,在无菌产品批签发时,重无菌检查结果,轻质量管理。
我国药典或相关法规应以适当方式提示至少注意以下几点:①由于无菌检查不可能对一批成品的每一包装单位进行检测,所以没有绝对的无菌保证;②无菌保证主要 由对灭菌过程 或 在CGMP下的无菌加工程序的验证来完成的,通过经验证的无菌检测方法提供支持;对于最终灭菌的产品提供灭菌过程的证据比无菌检查试验更重要;③无菌检查试验合格结果的保证水平还取决于批次的均一性、取样方法等的有效性;④即使整个批次均一,能检测出低水平污染的可能性也是非常小的;⑤由于不可能对每件产品检验, 就需要采取适 当的采样方案。
无菌检查方法的制订要考虑生物制品的特殊性
美国、欧洲、澳大利亚的药典或其它法规 中,对生物制品的无菌检查方法都另有专门的规定。我国2005 年版及之前版本药典中,生物制品的无菌检查方法不同于中药及化学药品,但是,2010年版实现了该方法在药典一、二、三 部的统一,使其与协调后的欧美日药典无菌检查法基本一致。生物制品因为其原料、生产过程、内含成份、具有生物活性等特点,使得产品中易于微生物特别是一些苛求微生物的存在,使用单一的无菌检查方法会存 在 风 险。我 国也应对生物制品的无菌检查做专门规定,特别在使用培养基和相应培养条件等方面。
抽样方案
抽样方案包括抽样数量和抽样方式,我国药典规定了不同药物产品的原液、半成品、成品最少抽验数量,没有对抽样方式做具体规定。参考欧洲、美国、澳大利亚的相关规定,药品无菌检查抽样方式应包含 如下几点:①无菌生产的产品在分装开始、中间和结束时抽样;在生 产中设备故障或异常、未经验证的中断或干扰情况发生后应及时抽样;②最终灭菌产品要在 灭菌装置的不同部位抽样,要包括灭菌因子最不易到达的位置;③用于重试的抽样应该考虑首次取样的位置和所处生产程序的时段。
对培养基的规定
中国药典无菌检查法规定使用2个培养基:硫乙醇酸盐流体培养基和改良马丁培养基,2015年版将实现与欧美日协调无菌检查法的统一,用胰酪胨大豆胨肉汤培养基替代改良马丁培养基。无论胰酪胨大豆胨肉汤培养基和改良马丁培养基二者哪一个更适用于无菌检查,用两个培养基完全覆盖所有需要无菌检查的药品并不现实。欧洲药典 “ 细胞产品微生物控制”没有指定培养基,但规定了更严格的培养基促生长性能检查法和基于实践中最可能污染微生物的方法验证。澳大利亚法规规定可用经验证的替代培养基检测可能的苛求微生物,推荐用停滞试验作为对培养基能力的附加验证方法。在中国药典三部2010年版,无菌检查法培养基灵敏度检查的测试菌株用金黄色葡萄球菌替代了2005年版的乙型溶血链球菌,削弱了对培养基品质的甄别能力(本实验室数据待发表)。因此,对于某些特殊药品应允许使用经验证的替代培养基。
作者:范文平,赵宏大,谢文, 中国食品药品检定研究院
来源:中国药事
本文仅为交流学习,版权归原作者所有,转载请注明出处。


![]()
2001-2009Vogel Industry Media版权所有 京ICP备12020067号-15 京公网安备110102001177号


加载更多